
Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Xeroderma pigmentosum
Expert medical al articolului

Ultima examinare: 04.07.2025
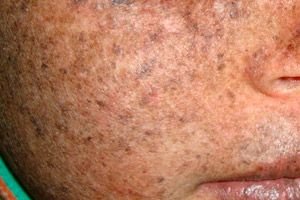
Xeroderma pigmentosum este o tulburare genetică autosomal recesivă a reparării ADN-ului. Boala este cauzată de mutații ale genelor implicate în repararea ADN-ului deteriorat de radiațiile ultraviolete și alte substanțe toxice.
Cel mai adesea, această boală afectează copiii, numiți și „copiii nopții”. Complicațiile frecvente ale bolii sunt carcinomul bazocelular și alte neoplasme maligne ale pielii, melanomul malign metastatic și carcinomul scuamos.
 [ 1 ]
[ 1 ]
Cauze xerodermie pigmentară
Patogeneza
Deficiența enzimelor endonucleaze UV în celulele pacientului (în fibroblaste) sau absența completă a acestora joacă un rol important în dezvoltarea bolii. Această enzimă este responsabilă de reproducerea ADN-ului deteriorat de razele ultraviolete. Conform altor surse, deficiența enzimei ADN polimerază-1 joacă un rol important în dezvoltarea bolii la unii pacienți. Boala se dezvoltă cel mai adesea sub influența razelor cu o lungime de undă de 280-310 nm. Se crede că xerodermia pigmentară se dezvoltă din cauza pătrunderii diferitelor substanțe fotodinamice în organism sau a creșterii porfirinelor în mediul biologic uman.
În stadiile incipiente ale bolii, se observă hiperkeratoză, atrofie a focarului epidermic, subțierea stratului malpighian, o creștere a granulelor de melanină în stratul bazal al celulelor și infiltrat inflamator cronic în stratul superior al dermului (în principal în jurul vaselor de sânge). Ulterior, se detectează modificări degenerative la nivelul colagenului și al fibrelor elastice, iar în stadiul tumoral, se dezvăluie semne caracteristice cancerului de piele.
Simptome xerodermie pigmentară
Bărbații și femeile sunt afectați în egală măsură de această boală. Boala începe în copilăria timpurie, primăvara sau vara. Pielea pacientului este deosebit de sensibilă la lumina soarelui. Prin urmare, prima erupție cutanată sub formă de hiperpigmentare, similară unei alunițe, apare pe zonele pielii expuse la lumina soarelui. Erupția se agravează, eritemul se intensifică, devine maro închis, apar telangiectazii, angiom și atrofie. Ulterior, cresc papiloame și negi (în principal pe pielea feței și a gâtului), apar pete și ulcere. Ca urmare a cicatricilor ulcerului, nasul devine mai subțire („ciocul păsării”), pleoapa se evertește. Papiloamele se dezvoltă de obicei în tumori maligne. Xerodermia pigmentată, de regulă, apare împreună cu cancerul spinocelular, melanosarcoame.
Ce te deranjează?
Etape
În evoluția clinică a xerodermei pigmentoase, se disting cinci etape: inflamatorie (eritematoasă), hiperpigmentare, atrofie, hiperkeratoză și tumori maligne.
În stadiul inflamator (eritematos), pe zonele de piele expuse la soare (față, gât, partea superioară a pieptului, brațe, mâini) apar umflături, pete roșii și uneori blistere și vezicule. Aproape că nu există erupție cutanată pe zonele neexpuse la soare.
În cazul hiperpigmentării, în locul petelor roșii apar pete hiperpigmentate de culoare maro deschis, asemănătoare alunițelor.
Odată cu atrofia, pielea se usucă, devine mai subțire și se încrețește. Pe pielea buzelor și a nasului se observă numeroase telangiectazii mici, stelate, și cicatrici cu o suprafață lucioasă. Din cauza atrofiei și a cicatricilor, se dezvoltă microtomie (reducerea deschiderii gurii), ectropion, subțierea urechilor și nasului, atrezie a deschiderii nazale și a gurii. La 80-85% dintre pacienți, ochii sunt afectați: se observă conjunctivită, keratită, leziuni ale corneei și mucoasei și slăbirea vederii. Pe pielea pleoapelor se observă discromie, telangiectazii, modificări hiperkeratotice și tumori.
În cazul hiperkeratozei, în focarele patologice descrise mai sus apar tumori verucoase, papiloame, keratoacantom, fibroame, angiofibriom și alte tumori benigne. Prin urmare, unii oameni de știință includ xerodermia pigmentară în grupul bolilor precanceroase.
După 10-15 ani de la debutul bolii, tumori maligne ale pielii (basaliom, endoteliom, angiosarcom) apar în focare și pete pigmentare alterate atrofic. Tumorile sunt distruse în scurt timp, metastazează la organele interne și duc la deces. În organele și țesuturile interne ale unor pacienți se observă modificări distrofice generale (sindactelia degetelor de la picior al doilea și al treilea, distrofie dentară, căderea completă a părului etc.).
Formulare
Forma neurologică a xerodermei pigmentoase se manifestă prin două sindroame.
Sindromul Reed este caracterizat prin manifestări clinice de xerodermă pigmentoasă și microcefalie, idiopatie și creștere lentă a sistemului osos. În forma neurologică a bolii, repararea ADN-ului este dificilă, iar radioterapia duce la o creștere a principalelor focare patologice.
Sindromul De Sinctis-X Cocchione se caracterizează prin următoarele semne clinice:
- dezvoltarea semnelor clinice de xerodermă pigmentoasă pe pielea deosebit de fotosensibilă;
- apariția precoce a tumorilor maligne;
- evoluție simultană a paraliziei spastice, microcefaliei și demenței;
- deformități congenitale și nanism;
- subdezvoltarea gonadelor;
- avorturi spontane frecvente;
- transmitere recesivă prin moștenire.
Potrivit unor dermatologi, sindromul Santis-Cacchione nu este o boală independentă, ci o manifestare clinică severă și complet exprimată a xerodermei pigmentoase.
Forme genetice
Tipuri |
Genă |
Locus |
Descriere |
Tip A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) grupa A – forma clasică |
Tip B, II, XPB |
XPB |
2q21 |
Grupa B XP |
Tip C, III, XPC |
XPC |
3p25 |
Grupa XP C |
Tip D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP Grupa D sau Sindromul De Sanctis-Cacchione |
Tip E, V, XPE |
DDB2 |
11p12-p11 |
Grupa XP E |
Tip F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
Grupa XP F |
Tip G, VII, XPG |
RAD2ERCC5 |
13q33 |
XP grup G și sindrom COFS (sindrom cerebro-oculo-facioscheletal) tip 3 |
Tip V, XPV |
POLH |
6p21.1-p12 |
Varianta Xeroderma Pigmentosa |
Ce trebuie să examinăm?
Cum să examinăm?
Cine să contactați?
Tratament xerodermie pigmentară
Medicamentele antimalarice (delagil, plaquenil, resoquin etc.), care protejează ADN-ul de razele ultraviolete, inhibă procesul de depolimerizare, reduc sensibilitatea (fotosensibilizarea) pielii la lumina soarelui. Aceste medicamente au proprietăți antiinflamatorii și hiposensibilizante. Se recomandă efectuarea terapiei generale împreună cu terapia cu vitamine (B1, B2, PP, B6, B12, A, E), antihistaminice (tavegil, difenhidramină, suprastin), agenți desensibilizanți (tiosulfat de sodiu, clorură de calciu 10%, intravenos, 10 ml).
Pentru tratamentul local, se folosesc creme și unguente cu protecție solară.
În forma tumorală de xerodermie pigmentară, se utilizează metode chirurgicale, azot lichid și raze laser. Pentru a vă proteja de lumina soarelui, trebuie să purtați haine largi, pălării de soare și mănuși.
Prognoză
Majoritatea pacienților (2/3) mor înainte de a împlini vârsta de 15 ani. Unii pacienți pot trăi până la 40-50 de ani.
[ 20 ]