
Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Nefrită ereditară (sindromul Alport) la copii
Expert medical al articolului

Ultima examinare: 05.07.2025
Nefrita ereditară (sindromul Alport) este o glomerulopatie ereditară, non-imună, determinată genetic, manifestată prin hematurie (uneori cu proteinurie), declin progresiv al funcției renale cu dezvoltarea insuficienței renale cronice, adesea combinată cu surditate neurosenzorială și deficiență de vedere.
Boala a fost descrisă pentru prima dată în 1902 de către LG Guthrie, care a observat o familie în care hematuria a fost observată în mai multe generații. În 1915, AF Hurst a descris dezvoltarea uremiei la membrii aceleiași familii. În 1927, A. Alport a identificat pentru prima dată pierderea auzului la mai multe rude cu hematurie. În anii 1950, au fost descrise leziuni oculare într-o boală similară. În 1972, la pacienții cu hematurie ereditară, în timpul unui studiu morfologic al țesutului renal, Hinglais și colab. au relevat o expansiune și o stratificare inegală a membranelor bazale glomerulare. În 1985, a fost identificată baza genetică a nefritei ereditare - o mutație în gena colagenului de tip IV (Fiengold și colab., 1985).
Studiul naturii genetice a bolii ne-a permis să concluzionăm că diferențele în manifestările fenotipice ale nefritei ereditare (cu sau fără pierderea auzului) se datorează gradului de exprimare a genei mutante. Astfel, în prezent, toate variantele clinice sunt considerate manifestări ale aceleiași boli, iar termenul „nefrită ereditară” este sinonim cu termenul „sindrom Alport”.
Conform studiilor epidemiologice, nefrita ereditară apare cu o frecvență de 17 la 100.000 de copii.
 [
[ Cauzele sindromului Alport
Baza genetică a bolii este o mutație în gena lanțului α-5 al colagenului de tip IV. Acest tip este universal pentru membranele bazale ale rinichiului, aparatul cohlear, capsula cristalinului, retină și cornee, fapt dovedit în studii care au utilizat anticorpi monoclonali împotriva acestei fracții de colagen. Recent, a fost indicată posibilitatea utilizării sondelor ADN pentru diagnosticul prenatal al nefritei ereditare.
Se subliniază importanța testării tuturor membrilor familiei cu sonde ADN pentru a identifica purtătorii genei mutante, ceea ce este de mare importanță în efectuarea consilierii medicale și genetice a familiilor cu această boală. Cu toate acestea, până la 20% din familii nu au rude care suferă de boli renale, ceea ce sugerează o frecvență ridicată a mutațiilor spontane ale genei anormale. Majoritatea pacienților cu nefrită ereditară au în familiile lor persoane cu boli renale, pierderea auzului și patologie a vederii; căsătoriile consangvine între persoane cu unul sau mai mulți strămoși sunt importante, deoarece în căsătoria persoanelor înrudite crește probabilitatea de a primi aceleași gene de la ambii părinți. Au fost stabilite căi de transmitere autosomal dominante, autosomal recesive și dominante, legate de cromozomul X.
La copii, cel mai frecvent se disting trei tipuri de nefrită ereditară: sindromul Alport, nefrita ereditară fără pierderea auzului și hematuria benignă familială.
Sindromul Alport este o nefrită ereditară cu deficiență de auz. Se bazează pe un defect combinat în structura colagenului membranei bazale glomerulare a rinichilor, structurilor urechii și ochiului. Gena sindromului Alport clasic este localizată în locusul 21-22 q al brațului lung al cromozomului X. În majoritatea cazurilor, este moștenit dominant, legat de cromozomul X. În acest sens, sindromul Alport este mai sever la bărbați, deoarece la femei funcția genei mutante este compensată de o alelă sănătoasă a celui de-al doilea cromozom, nedeteriorat.
Baza genetică pentru dezvoltarea nefritei ereditare o reprezintă mutațiile din genele lanțurilor alfa ale colagenului de tip IV. Sunt cunoscute șase lanțuri alfa ale colagenului G de tip IV: genele lanțurilor a5 și a6 (Col4A5 și Col4A5) sunt situate pe brațul lung al cromozomului X în zona 21-22q; genele lanțurilor a3 și a4 (Col4A3 și Col4A4) sunt situate pe cromozomul 2; genele lanțurilor a1 și a2 (Col4A1 și Col4A2) sunt situate pe cromozomul 13.
În majoritatea cazurilor (80-85%), se detectează un model de moștenire legat de cromozomul X al bolii, asociat cu deteriorarea genei Col4A5 ca urmare a deleției, mutațiilor punctuale sau tulburărilor de splicing. În prezent, au fost descoperite peste 200 de mutații ale genei Col4A5, responsabile de perturbarea sintezei lanțurilor a5 ale colagenului de tip IV. Cu acest tip de moștenire, boala se manifestă la copiii de ambele sexe, dar la băieți este mai severă.
Mutațiile în loci-urile genelor Col4A3 și Col4A4 responsabile de sinteza lanțurilor α3 și α4 ale colagenului de tip IV sunt moștenite autozomal. Conform cercetărilor, tipul autosomal dominant de moștenire este observat în 16% din cazurile de nefrită ereditară, iar tipul autosomal recesiv este observat la 6% dintre pacienți. Sunt cunoscute aproximativ 10 variante de mutații ale genelor Col4A3 și Col4A4.
Rezultatul mutațiilor este o încălcare a proceselor de asamblare a colagenului de tip IV, ceea ce duce la o încălcare a structurii sale. Colagenul de tip IV este una dintre principalele componente ale membranei bazale glomerulare, aparatului cohlear și cristalinului, a cărui patologie va fi detectată în clinica nefritei ereditare.
Colagenul de tip IV, care face parte din membrana bazală glomerulară, este alcătuit în principal din două lanțuri α1 (IV) și un lanț α2 (IV) și conține, de asemenea, lanțuri α3, α4, α5. Cel mai adesea, în moștenirea legată de cromozomul X, mutația genei Col4A5 este însoțită de absența lanțurilor α3, α4, α5 și α6 din structura colagenului de tip IV, iar numărul de lanțuri α1 și α2 din membrana bazală glomerulară crește. Mecanismul acestui fenomen este neclar, presupunându-se că cauza o reprezintă modificările post-transcripționale ale ARNm.
Absența lanțurilor α3, α4 și α5 din structura colagenului de tip IV al membranelor bazale glomerulare duce la subțierea și fragilitatea acestora în stadiile incipiente ale sindromului Alport, manifestată clinic mai des prin hematurie (mai rar prin hematurie cu proteinurie sau doar proteinurie), pierderea auzului și lenticonus. Progresia ulterioară a bolii duce la îngroșarea și afectarea permeabilității membranelor bazale în stadiile avansate ale bolii, cu proliferarea în acestea a colagenului de tip V și VI, manifestată printr-o creștere a proteinuriei și o scădere a funcției renale.
Natura mutației care stă la baza nefritei ereditare determină în mare măsură manifestarea sa fenotipică. În cazul deleției cromozomului X cu mutația simultană a genelor Col4A5 și Col4A6, responsabile de sinteza lanțurilor α5 și α6 ale colagenului de tip IV, sindromul Alport este combinat cu leiomiomatoza esofagiană și a organelor genitale. Conform datelor cercetărilor, în cazul unei mutații a genei Col4A5 asociate cu o deleție, se observă o severitate mai mare a procesului patologic, o combinație de afectare renală cu manifestări extrarenale și dezvoltarea precoce a insuficienței renale cronice, comparativ cu o mutație punctuală a acestei gene.
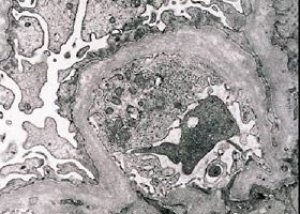
Din punct de vedere morfologic, microscopia electronică relevă subțierea și stratificarea membranelor bazale glomerulare (în special a laminei densa) și prezența granulelor electron-dense. Leziunile glomerulare pot fi eterogene la același pacient, de la leziuni mezangiale focale minime până la glomeruloscleroză. Glomerulita în sindromul Alport este întotdeauna imunonegativă, ceea ce o distinge de glomerulonefrită. Caracteristicile includ dezvoltarea atrofiei tubulare, infiltrarea limfohistiocitară și prezența „celulelor spumoase” cu incluziuni lipidice - lipofagi. Pe măsură ce boala progresează, se evidențiază îngroșarea și distrugerea pronunțată a membranelor bazale glomerulare.
Sunt evidențiate anumite modificări ale sistemului imunitar. Pacienții cu nefrită ereditară au un nivel scăzut de Ig A și o tendință de creștere a concentrației de IgM în sânge, nivelul de IgG putând fi crescut în stadiile incipiente ale bolii și scăzut în stadiile ulterioare. Este posibil ca creșterea concentrației de IgM și G să fie un fel de reacție compensatorie ca răspuns la deficitul de IgA.
Activitatea funcțională a sistemului limfocitar T este redusă; se observă o scădere selectivă a limfocitelor B responsabile de sinteza Ig A, legătura fagocitară a imunității este perturbată, în principal din cauza perturbării chemotaxiei și a proceselor de digestie intracelulară în neutrofile.
La examinarea unei biopsii renale la pacienții cu sindrom Alport, datele de microscopie electronică relevă modificări ultrastructurale ale membranei bazale glomerulare: subțierea, perturbarea structurii și divizarea membranelor bazale glomerulare cu o modificare a grosimii și contururi neuniforme. În stadiile incipiente ale nefritei ereditare, defectul determină subțierea și fragilitatea membranelor bazale glomerulare.
Subțierea membranelor glomerulare este un semn mai favorabil și este mai frecventă la fete. Un semn microscopic electronic mai constant în nefrita ereditară este divizarea membranei bazale, iar severitatea distrugerii acesteia se corelează cu severitatea procesului.
Simptomele sindromului Alport la copii
Primele simptome ale sindromului Alport, sub forma unui sindrom urinar izolat, sunt cel mai adesea detectate la copiii din primii trei ani de viață. În majoritatea cazurilor, boala este detectată întâmplător. Sindromul urinar este detectat în timpul unui examen preventiv al copilului, înainte de internarea într-o creșă sau în timpul infecțiilor respiratorii acute (AVI). În cazul apariției unor patologii în urină în timpul AVI. În nefrita ereditară, spre deosebire de glomerulonefrita dobândită, nu există o perioadă latentă.
În stadiul inițial al bolii, sănătatea copilului are puțin de suferit, o trăsătură caracteristică fiind persistența și rezistența sindromului urinar. Unul dintre principalele semne este hematuria de diferite grade de severitate, observată în 100% din cazuri. O creștere a gradului de hematurie se observă în timpul sau după infecțiile respiratorii, activitatea fizică sau după vaccinările preventive. Proteinuria în majoritatea cazurilor nu depășește 1 g/zi, la începutul bolii poate fi inconstantă, pe măsură ce procesul progresează, proteinuria crește. Periodic, în sedimentul urinar poate fi prezentă leucociturie cu predominanță de limfocite, ceea ce este asociat cu dezvoltarea modificărilor interstițiale.
Ulterior, funcția renală parțială este afectată, starea generală a pacientului se agravează: apar intoxicație, slăbiciune musculară, hipotensiune arterială, adesea deficit de auz (în special la băieți) și uneori deficit de vedere. Intoxicația se manifestă prin paloare, oboseală și dureri de cap. În stadiul inițial al bolii, pierderea auzului este în majoritatea cazurilor detectată doar prin audiografie. Pierderea auzului în sindromul Alport poate apărea în diferite perioade ale copilăriei, dar cel mai adesea pierderea auzului este diagnosticată la vârsta de 6-10 ani. Pierderea auzului la copii începe cu frecvențe înalte, atingând un grad semnificativ în conducerea aeriană și osoasă, trecând de la pierderea auzului prin conducerea sunetului la pierderea auzului prin perceperea sunetului. Pierderea auzului poate fi unul dintre primele simptome ale bolii și poate preceda sindromul urinar.
În 20% din cazuri, pacienții cu sindrom Alport prezintă modificări ale organelor vizuale. Cele mai frecvent detectate anomalii sunt cele ale cristalinului: sferofokie, lenticon anterior, posterior sau mixt și diverse cataracte. În familiile cu sindrom Alport, există o frecvență semnificativă a miopiei. O serie de cercetători observă constant modificări perimaculare bilaterale în aceste familii sub forma unor granulații albicioase sau gălbui strălucitoare în corpul galben. Aceștia consideră acest semn ca fiind un simptom constant care are o valoare diagnostică ridicată în sindromul Alport. K. S. Chugh și colab. (1993) într-un studiu oftalmologic au constatat la pacienții cu sindrom Alport o scădere a acuității vizuale în 66,7% din cazuri, lenticon anterior în 37,8%, pete retiniene în 22,2%, cataractă în 20% și keratocon în 6,7%.
La unii copii cu nefrită ereditară, în special atunci când se dezvoltă insuficiență renală, se observă o întârziere semnificativă în dezvoltarea fizică. Pe măsură ce insuficiența renală progresează, se dezvoltă hipertensiunea arterială. La copii, aceasta este mai des detectată în adolescență și la grupele de vârstă mai înaintată.
Pacienții cu nefrită ereditară se caracterizează prin prezența diferitelor (mai mult de 5-7) stigmate de dismorfogeneză a țesutului conjunctiv. Printre stigmatele țesutului conjunctiv la pacienți, cele mai frecvente sunt hipertelorismul ochilor, palatul înalt, anomaliile mușcăturii, forma anormală a auriculelor, curbura degetului mic la mâini și „spațiul de sandală” la picioare. Nefrita ereditară se caracterizează prin uniformitatea stigmatelor de dismorfogeneză în cadrul unei familii, precum și prin frecvența ridicată a distribuției acestora între rudele probandilor de-a lungul cărora se transmite boala.
În stadiile incipiente ale bolii, se detectează o scădere izolată a funcțiilor renale parțiale: transportul aminoacizilor, electroliților, funcția de concentrare, acidogeneza, modificările ulterioare afectează starea funcțională atât a părții proximale, cât și a celei distale a nefronului și se caracterizează prin tulburări parțiale combinate. O scădere a filtrării glomerulare apare mai târziu, mai des în adolescență. Pe măsură ce nefrita ereditară progresează, se dezvoltă anemia.
Astfel, nefrita ereditară se caracterizează printr-o evoluție stadială a bolii: mai întâi, un stadiu latent sau simptome clinice ascunse, manifestate prin modificări minime ale sindromului urinar, apoi are loc o decompensare treptată a procesului cu o scădere a funcției renale cu simptome clinice manifeste (intoxicație, astenie, întârziere de dezvoltare, anemie). Simptomele clinice apar de obicei indiferent de stratificarea reacției inflamatorii.
Nefrita ereditară se poate manifesta la diferite perioade de vârstă, ceea ce depinde de acțiunea genei care se află într-o stare reprimată până la un anumit moment.
Clasificare
Există trei tipuri de nefrită ereditară
- Opțiunea I - se manifestă clinic ca nefrită cu hematurie, pierderea auzului și leziuni oculare. Cursul nefritei este progresiv odată cu dezvoltarea insuficienței renale cronice. Tipul de moștenire este dominant, legat de cromozomul X. Morfologic, se evidențiază o încălcare a structurii membranei bazale, subțierea și divizarea acesteia.
- Opțiunea II - se manifestă clinic ca nefrită cu hematurie fără pierderea auzului. Evoluția nefritei este progresivă odată cu dezvoltarea insuficienței renale cronice. Tipul de moștenire este dominant, legat de cromozomul X. Morfologic, se detectează subțierea membranei bazale a capilarelor glomerulare (în special a laminadensei).
- Opțiunea III - hematurie familială benignă. Evoluția este favorabilă, nu se dezvoltă insuficiență renală cronică. Tipul de moștenire este autosomal dominant sau autosomal recesiv. În cazul tipului de moștenire autosomal recesiv, la femei se observă o evoluție mai severă a bolii.
Diagnosticul sindromului Alport
Se propun următoarele criterii:
- prezența a cel puțin doi pacienți cu nefropatie în fiecare familie;
- hematurie ca principal simptom al nefropatiei la proband;
- prezența pierderii auzului la cel puțin un membru al familiei;
- dezvoltarea insuficienței renale cronice la una sau mai multe rude.
În diagnosticarea diferitelor boli ereditare și congenitale, un loc important este acordat unei abordări cuprinzătoare a examinării și, mai presus de toate, atenției acordate datelor obținute la întocmirea pedigree-ului copilului. Diagnosticul sindromului Alport este considerat valid în cazurile în care la pacient sunt detectate 3 din 4 semne tipice: prezența hematuriei și insuficienței renale cronice în familie, prezența pierderii auzului neurosenzoriale, patologia vederii la pacient, detectarea semnelor de scindare a membranei bazale glomerulare cu o modificare a grosimii acesteia și contururi neuniforme în timpul caracteristicilor microscopice electronice ale biopsiei.
Examinarea pacientului trebuie să includă metode de cercetare clinică și genetică; studiul țintit al istoricului bolii; examinarea generală a pacientului, luând în considerare criterii semnificative din punct de vedere diagnostic. În stadiul de compensare, patologia poate fi detectată doar concentrându-se pe sindroame precum prezența unei poveri ereditare, hipotensiune arterială, stigmate multiple de disembriogeneză, modificări ale sindromului urinar. În stadiul de decompensare, pot apărea simptome extrarenale, cum ar fi intoxicația severă, astenia, dezvoltarea fizică întârziată, anemia, manifestându-se și intensificându-se cu o scădere treptată a funcției renale. La majoritatea pacienților, cu o scădere a funcției renale, se observă următoarele: scăderea acido- și aminogenezei; 50% dintre pacienți observă o scădere semnificativă a funcției secretorii a rinichilor; gamă limitată de fluctuații ale densității optice a urinei; perturbarea ritmului de filtrare și apoi o scădere a filtrării glomerulare. Stadiul insuficienței renale cronice este diagnosticat atunci când pacienții prezintă un nivel crescut de uree în serul sanguin (mai mult de 0,35 g/l) timp de 3-6 luni sau mai mult și o scădere a filtrării glomerulare la 25% din normă.
Diagnosticul diferențial al nefritei ereditare trebuie efectuat în primul rând cu forma hematurică a glomerulonefritei dobândite. Glomerulonefrita dobândită are cel mai adesea un debut acut, o perioadă de 2-3 săptămâni după infecție, semne extrarenale, inclusiv hipertensiune arterială din primele zile (în nefrita ereditară, dimpotrivă, hipotensiune arterială), scăderea filtrării glomerulare la debutul bolii, fără afectarea funcțiilor tubulare parțiale, în timp ce în ereditară acestea sunt prezente. Glomerulonefrita dobândită apare cu hematurie și proteinurie mai pronunțate, cu o VSH crescută. Modificările tipice ale membranei bazale glomerulare, caracteristice nefritei ereditare, au valoare diagnostică.
Diagnosticul diferențial al nefropatiei dismetabolice se efectuează cu insuficiența renală cronică, în cazul bolilor renale eterogene manifestate clinic în familie, putând exista un spectru de nefropatie de la pielonefrită la urolitiază. Copiii au adesea plângeri de dureri abdominale și, periodic, în timpul urinării, în sedimentul urinar - oxalați.
Dacă se suspectează nefrită ereditară, pacientul trebuie trimis la un departament specializat de nefrologie pentru clarificarea diagnosticului.
Ce trebuie să examinăm?
Cum să examinăm?
Ce teste sunt necesare?
Cine să contactați?
Tratamentul sindromului Alport
Regimul include restricții privind efortul fizic intens și expunerea la aer proaspăt. Dieta este completă, cu niveluri suficiente de proteine, grăsimi și carbohidrați completi, ținând cont de funcția renală. De mare importanță este detectarea și tratamentul focarelor cronice de infecție. Se utilizează următoarele medicamente: ATP, cocarboxilază, piridoxină (până la 50 mg/zi), clorură de carnitină. Curele se administrează de 2-3 ori pe an. Pentru hematurie se prescriu medicamente pe bază de plante - urzică, suc de aronia, coada-șoricelului.
Există rapoarte în literatura de specialitate, atât străină, cât și internă, despre tratamentul cu prednisolon și utilizarea citostaticelor. Cu toate acestea, este dificil de evaluat efectul.
În insuficiența renală cronică se utilizează hemodializa și transplantul de rinichi.
Nu există metode de terapie specifică (patogenetică eficientă) pentru nefrita ereditară. Toate măsurile de tratament vizează prevenirea și încetinirea declinului funcției renale.
Dieta trebuie să fie echilibrată și hipercalorică, ținând cont de starea funcțională a rinichilor. În absența tulburărilor funcționale, dieta copilului trebuie să conțină suficiente proteine, grăsimi și carbohidrați. În prezența semnelor de disfuncție renală, cantitatea de proteine, carbohidrați, calciu și fosfor trebuie limitată, ceea ce întârzie dezvoltarea insuficienței renale cronice.
Activitatea fizică trebuie limitată; copiilor li se recomandă să evite sporturile.
Contactul cu pacienții infecțioși trebuie evitat, riscul de a dezvolta boli respiratorii acute trebuie redus. Este necesară igienizarea focarelor de infecție cronică. Vaccinările preventive nu se efectuează la copiii cu nefrită ereditară, vaccinarea este posibilă doar pentru indicații epidemiologice.
Terapia hormonală și imunosupresoare în nefrita ereditară este ineficientă. Există indicii ale unui anumit efect pozitiv (reducerea proteinuriei și încetinirea progresiei bolii) în cazul utilizării pe termen lung, timp de mai mulți ani, a ciclosporinei A și a inhibitorilor ECA.
În tratamentul pacienților, se utilizează medicamente care îmbunătățesc metabolismul:
- piridoxină - 2-3 mg/kg/zi în 3 doze timp de 4 săptămâni;
- cocarboxilază - 50 mg intramuscular o dată la două zile, un total de 10-15 injecții;
- ATP - 1 ml intramuscular o dată la două zile, 10-15 injecții;
- vitamina A - 1000 UI/an/zi într-o singură doză timp de 2 săptămâni;
- Vitamina E - 1 mg/kg/zi într-o singură doză, timp de 2 săptămâni.
Acest tip de terapie ajută la îmbunătățirea stării generale a pacienților, la reducerea disfuncțiilor tubulare și se desfășoară în cure de 3 ori pe an.
Levamisolul poate fi utilizat ca imunomodulator - 2 mg/kg/zi de 2-3 ori pe săptămână, cu pauze între doze de 3-4 zile.
Conform datelor cercetărilor, oxigenarea hiperbarică are un efect pozitiv asupra severității hematuriei și a disfuncției renale.
Cea mai eficientă metodă de tratare a nefritei ereditare este transplantul renal efectuat la timp. În acest caz, nu există recidivă a bolii în timpul transplantului; într-un procent mic de cazuri (aproximativ 5%), nefrita se poate dezvolta în rinichiul transplantat, asociată cu antigene ale membranei bazale glomerulare.
O direcție promițătoare este diagnosticul prenatal și terapia prin inginerie genetică. Experimentele pe animale arată o eficiență ridicată a transferului genelor normale responsabile de sinteza lanțurilor alfa de colagen de tip IV în țesutul renal, după care se observă sinteza structurilor normale de colagen.
Prognoză
Prognosticul nefritei ereditare este întotdeauna grav.
Criteriile prognostice nefavorabile pentru evoluția nefritei ereditare sunt:
- sex masculin;
- dezvoltarea precoce a insuficienței renale cronice la membrii familiei;
- proteinurie (mai mult de 1 g/zi);
- îngroșarea membranelor bazale glomerulare conform microscopiei;
- neurită acustică;
- deleție în gena Col4A5.
Prognosticul pentru hematuria familială benignă este mai favorabil.