
Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Sindromul Pierre Robin
Expert medical al articolului

Ultima examinare: 04.07.2025

Sindromul Pierre Robin, cunoscut și în medicină sub numele de anomalie Robin, este o patologie congenitală a dezvoltării părții maxilarului a feței. Boala și-a primit numele în onoarea dentistului francez P. Robin, care a descris pentru prima dată toate semnele sale. Lannelongue și Menard au descris pentru prima dată sindromul Pierre Robin în 1891, în raportul lor despre doi pacienți cu micrognație, palatoschizis și retroglossoptoză. În 1926, Pierre-Robin a publicat un caz al bolii la un sugar cu semne ale sindromului clasic. Până în 1974, triada de semne era cunoscută sub numele de sindromul Robin-Pierre. Cu toate acestea, acest sindrom este folosit acum pentru a descrie malformații cu prezența simultană a mai multor anomalii.
Epidemiologie
Este un defect congenital eterogen cu o prevalență de 1 la 8.500 de nou-născuți vii. Raportul dintre bărbați și femei este de 1:1, cu excepția formei legate de cromozomul X.
Dintre acești pacienți, 50% dintre sugari au o fisură palatină incompletă, restul se nasc cu un palat arcuit și neobișnuit de înalt, dar fără fisură.
Cauze Sindromul Pierre Robin
Se ia în considerare posibilitatea moștenirii autosomal recesive a bolii. Există două tipuri de sindrom, în funcție de etiologie: izolat și determinat genetic. Tipul izolat se dezvoltă din cauza compresiei părții inferioare a maxilarului în timpul dezvoltării embrionare. Compresia se poate dezvolta din cauza:
- Prezența sigiliilor localizate în uter (chisturi, cicatrici, tumori).
- Sarcină multiplă.
De asemenea, dezvoltarea maxilarului la făt poate fi perturbată de:
- Infecții virale pe care viitoarea mamă le-a suferit în timpul sarcinii.
- Tulburări neurotrofice.
- Cantitate insuficientă de acid folic în corpul unei femei însărcinate.
Patogeneza
Sindromul Pierre Robin este cauzat de tulburări embrionare care sunt cauzate de o mare varietate de patologii în perioada prenatală.
Există trei teorii fiziopatologice care ar putea explica apariția sindromului Pierre Robin.
Teoria mecanică: Această teorie este cea mai probabilă. Subdezvoltarea aparatului mandibular are loc între săptămânile 7 și 11 de sarcină. Poziția înaltă a limbii în cavitatea bucală duce la formarea de fisuri la nivelul palatului, din cauza cărora vena cavă nu se închide. Această teorie explică fisura clasică în formă de U inversat și absența fisurii labiale asociate. Oligohidramniosul poate juca un rol în etiologie, deoarece absența lichidului amniotic poate duce la deformarea bărbiei și compresia ulterioară a limbii între venele cave.
Teoria neurologică: O întârziere în dezvoltarea neurologică a fost observată prin electromiografia mușchilor uvulei și ai coloanei faringiene, iar gustul este cauzat de întârzierea conducerii în nervul hipoglos.
Teoria disneuroreglării rombencefalului: Această teorie se bazează pe perturbarea dezvoltării rombencefalului în timpul ontogenezei.
Dezvoltarea insuficientă a părții inferioare a maxilarului copilului duce la reducerea semnificativă a cavității bucale. Aceasta, la rândul său, provoacă așa-numita pseudomacroglosie, adică limba este deplasată în spatele peretelui faringian. Această patologie duce la dezvoltarea obstrucției căilor respiratorii.
Atâta timp cât bebelușul plânge sau se mișcă, căile respiratorii rămân libere, dar imediat ce bebelușul adoarme, obstrucția apare din nou.
Din cauza tulburărilor respiratorii, procesul de hrănire a bebelușului este foarte dificil. În acest moment, obstrucția căilor respiratorii apare aproape întotdeauna. Dacă nu se aplică nicio corecție medicală, o astfel de patologie poate duce la epuizare severă a întregului corp și chiar la moarte.
Simptome Sindromul Pierre Robin
Boala se caracterizează prin trei simptome principale:
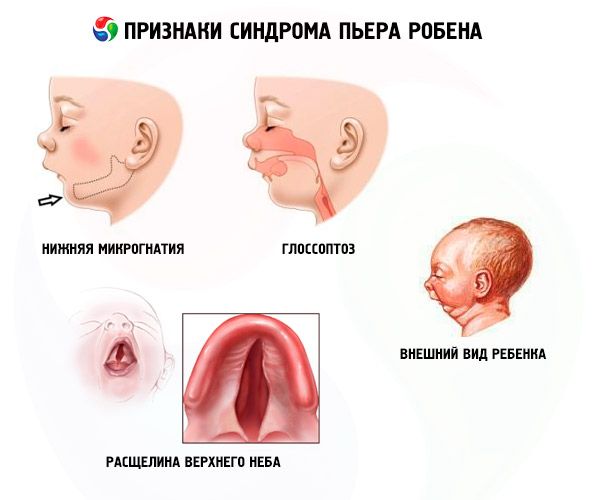
- Micrognația inferioară (subdezvoltarea maxilarului inferior, apare în 91,7% din cazurile de boală). Se caracterizează prin retracția arcadei dentare inferioare cu 10-12 mm în spatele arcadei superioare. Maxilarul inferior are un corp mic, un unghi obtuz. Copilul atinge o dezvoltare normală la aproximativ 5-6 ani.
- Glosoptoză (retracția limbii din cauza dezvoltării sale insuficiente, observată în 70-85% din cazuri).
- Macroglosia și anchiloglosia sunt simptome relativ rare, observate în 10-15% din cazuri.
- O crăpătură apare pe cer.
- Bradipnee și dispnee.
- Cianoză ușoară.
- Asfixie, care apare cel mai adesea în timpul încercărilor de a hrăni copilul.
- Înghițirea este imposibilă sau foarte dificilă.
- Senzație de vomă.
- Anomalii auriculare în 75% din cazuri.
- Hipoacuzia conductivă apare la 60% dintre pacienți, în timp ce atrezia canalului auditiv extern apare doar la 5% dintre pacienți, pneumatizarea insuficientă a cavității mastoide a osului temporal.
- Anomalii ale urechii interne (aplazia canalelor semicirculare laterale, apeductul vestibular mare, pierderea celulelor ciliate cohleare).
- Malformațiile nazale sunt mai puțin frecvente și constau în principal în anomalii ale rădăcinii nazale.
- Malformațiile dentare apar în 30% din cazuri. Laringomalacia și insuficiența velofaringiană apar la aproximativ 10-15% dintre pacienții cu sindrom Pierre Robin.
Caracteristici sistemice ale sindromului Pierre Robin
Anomaliile sistemice de dezvoltare sunt descrise în 10-85% din cazurile înregistrate.
Anomaliile oculare apar la 10-30% dintre pacienți. Acestea pot include: hipermetropie, miopie, astigmatism, scleroză corneană și stenoză a canalului nazolacrimal.
Patologii cardiovasculare: sufluri benigne la inimă, stenoză a arterei pulmonare, canal arterial permeabil, fereastră ovală, defectul septal atrial și hipertensiune pulmonară. Prevalența acestora variază între 5-58%.
Anomalii legate de sistemul musculo-scheletic (70-80% din cazuri): sindactilie, falange displazice, polidactilie, clinodactilie, hipermobilitatea articulațiilor și oligodactilie a membrelor superioare. Anomalii ale membrelor inferioare: anomalii ale piciorului (picior strâmb, adducție metatarsiană), malformații femurale (pelvis valgus sau varus, femur scurt), anomalii ale șoldului (luxații congenitale, contracturi), anomalii ale articulației genunchiului (GENU VALGUS, sincondroză). Malformații ale coloanei vertebrale: scolioză, cifoză, lordoză, displazie vertebrală, agenezie a sacrului și sinusului coccigian.
Patologia sistemului nervos central: epilepsie, întârzieri în dezvoltarea sistemului nervos, hidrocefalie. Frecvența defectelor SNC este de aproximativ 50%.
Anomalii genito-urinare: testicule necoborate (25%), hidronefroză (15%) și hidrocel (10%).
Sindroame și afecțiuni asociate: sindromul Stickler, sindromul trisomiei 11q, trisomia 18, sindromul deleției 4q, artropatia reumatoidă, hipocondroplazia, sindromul Moebius.
Etape
Există trei stadii de severitate a bolii, care depind de starea tractului respirator al copilului:
- Ușoară - există probleme minore cu hrănirea, dar respirația este aproape lipsită de dificultăți. Tratamentul se efectuează în regim ambulatoriu.
- Moderat – respirația este moderat dificilă, hrănirea copilului este moderat dificilă. Tratamentul se efectuează în spital.
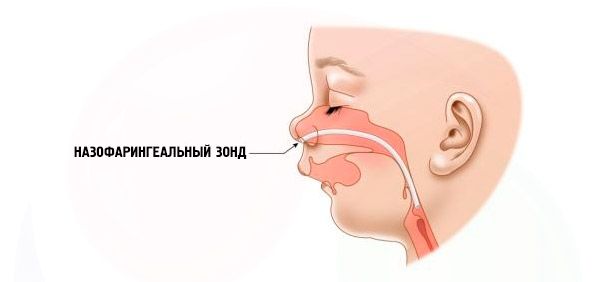
- Severă – respirația este foarte dificilă, copilul nu poate fi hrănit normal. Este necesară utilizarea unor dispozitive speciale (sondă intranazală).
Complicații și consecințe
Combinația dintre micrognație și glosoptoză poate duce la complicații respiratorii severe și probleme în timpul hrănirii copilului.
Sindromul Pierre Robin provoacă următoarele complicații:
- Respirație stridoză din cauza obstrucției căilor respiratorii. Laringomalacie sau chiar asfixie în somn.
- Dezvoltarea psihomotorie a copilului este mult în urma colegilor săi.
- Dezvoltarea fizică este, de asemenea, în urmă.
- Vorbirea pacienților este afectată.
- Infecții frecvente ale urechii care devin cronice și duc la deficiențe de auz.
- Sindromul de apnee obstructivă în somn, apariția decesului în somn variază în 14-91% din cazuri.
- Probleme cu dinții.
Diagnostice Sindromul Pierre Robin
Diagnosticul sindromului Pierre Robin nu este dificil. Se bazează pe manifestările clinice. Pentru a exclude alte patologii, este foarte important să consultați un genetician.
Copiii cu anomalie congenitală Robin au probleme respiratorii de la naștere din cauza limbii care se afundă constant înapoi. Bebelușul este neliniștit, pielea lui este albăstruie, respirația șuierătoare iese din piept la inhalare. Se poate produce sufocare în timpul hrănirii. Diagnosticul poate fi pus și pe baza aspectului neobișnuit al copilului - „față de pasăre”. Adesea, pacienții dezvoltă și alte defecte: miopie, cataractă, patologie a sistemului genitourinar, patologie cardiacă, anomalii în dezvoltarea coloanei vertebrale.
Pe baza acestor manifestări clinice, nu va fi dificil pentru un specialist să pună un diagnostic corect.
Cine să contactați?
Tratament Sindromul Pierre Robin
Tratamentul se efectuează imediat după nașterea unui copil cu sindrom Pierre Robin. Dacă boala este ușoară, atunci pentru a îmbunătăți starea pacientului este necesar să se țină constant copilul vertical sau culcat pe burtă. Capul bebelușului trebuie înclinat spre piept. În timpul hrănirii, nu se recomandă ținerea copilului în poziție orizontală, astfel încât alimentele să nu ajungă în tractul respirator.
Dacă subdezvoltarea maxilarului inferior este destul de pronunțată, se recurge la intervenția chirurgicală pentru a aduce limba retrasă într-o poziție fiziologică normală. În cazurile severe, limba este trasă în sus și fixată pe buza inferioară. În cazurile foarte severe, trebuie efectuată traheostomie, glossopexie și osteogeneză prin distragere a maxilarului inferior.
Se utilizează și tratamentul conservator.
Medicamente
Fenobarbital. Un somnifer și un medicament sedativ, are efect anticonvulsivant. Fiecare comprimat conține 100 ml de fenobarbital. Dozajul este individual, deoarece depinde de severitatea bolii și de starea copilului. Medicamentul este interzis pacienților cu insuficiență hepatică, hiperkinezie, anemie, miastenie, porfirie, diabet zaharat, depresie și intoleranță la componente. Următoarele simptome sunt posibile în timpul administrării: amețeli, astenie, halucinații, agranulocitoză, greață, tensiune arterială scăzută și alergii.
Clonazepam. Un medicament prescris pentru tratamentul epilepsiei. Medicamentul conține substanța activă clonazepam, care este un derivat de benzodiazepină. Are efecte anticonvulsivante, anxiolitice și miorelaxante. Doza este stabilită de medicul curant, dar nu trebuie să depășească doza maximă - 250 mcg pe zi. Nu luați în caz de insomnie, hipertonie musculară, agitație psihomotorie, tulburări de panică. Următoarele simptome sunt posibile în timpul administrării: letargie, greață, dismenoree, cefalee, leucopenie, retenție urinară sau incontinență, alopecie, alergie.
Sibazon. Disponibil sub formă de soluție și comprimate rectale. Substanța activă este un derivat de benzodiazepină (sibazon). Are efect sedativ, anxiolitic, anticonvulsivant. Dozajul este individual. Pacienților cu hipercapnie cronică, miastenie, intoleranță la benzodiazepine le este interzis să ia medicamentul. În timpul utilizării medicamentului, pot apărea următoarele simptome: greață, constipație, cefalee, amețeli, sughiț, incontinență urinară, alergii.
Liofilizat de cortexină. Un medicament cu efect nootrop. Medicamentul conține un complex de fracțiuni polipeptidice solubile în apă și glicină. Dozajul este individual și este prescris de medicul curant în funcție de starea pacientului. Pacienților cu intoleranță la cortexină le este interzis să ia acest medicament. Medicamentul poate provoca reacții alergice.
Tratament de fizioterapie
De obicei, în stadiile ușoare ale sindromului, se utilizează terapia pozițională, în care copilul este plasat pe burtă în poziție verticală până când gravitația forțează maxilarul inferior să crească corect.
Tratament chirurgical
Tratamentul chirurgical este utilizat în principal pentru corectarea glosoptozei. Există mai multe metode:
- Susținerea limbii cu un fir de argint. Firul este trecut prin partea inferioară a gingiei și buza inferioară. Metoda se numește Douglas.
- Metoda Duhamel - un fir gros de argint este trecut prin baza limbii pacientului și prin ambii obraji. Se utilizează nu mai mult de treizeci de zile.
- Dispozitive ortopedice pentru extensia și fixarea limbii.
- La vârsta de un an, se poate efectua o intervenție chirurgicală pentru corectarea palatoschizisului.
Prognoză
Prognosticul și evoluția bolii sunt severe. Cel mai adesea, decesul survine în primele zile de viață, în stadiul moderat și sever al bolii (cauza fiind asfixia). De asemenea, riscul de deces în primul an este destul de mare din cauza numeroaselor infecții.
Pentru pacienții cu vârsta peste doi ani, prognosticul este favorabil.
 [ 36 ]
[ 36 ]