
Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Sindromul Treacher Collins
Expert medical al articolului

Ultima examinare: 04.07.2025

Tulburările intrauterine ale proceselor de dezvoltare osoasă provoacă deformări craniofaciale grave, iar una dintre varietățile unei astfel de patologii este sindromul Treacher Collins (TCS) sau mandibulofascial, adică disostoza maxilofacială.
Codul bolii conform ICD 10: clasa XVII (anomalii congenitale, deformări și tulburări cromozomiale), Q75.4 - disostoză mandibulofacială.
Cauze Sindromul Treacher Collins
Acest sindrom a fost numit după remarcabilul oftalmolog britanic Edward Treacher Collins, care a descris principalele caracteristici ale patologiei în urmă cu mai bine de o sută de ani. Cu toate acestea, medicii europeni numesc mai des acest tip de anomalie a oaselor faciale și maxilare boala sau sindromul Franceschetti - pe baza cercetărilor ample ale oftalmologului elvețian Adolf Franceschetti, care a introdus termenul de „disostoză mandibulofascială” la mijlocul secolului trecut. În cercurile medicale, se folosește și denumirea de sindrom Franceschetti-Collins.
Sindromul Treacher Collins este cauzat de mutații ale genei TCOF1 (la locusul cromozomului 5q31.3-33.3), care codifică o fosfoproteină nucleolară responsabilă de formarea părții craniofaciale a embrionului uman. Ca urmare a unei scăderi premature a cantității acestei proteine, biogeneza și funcțiile ARNr sunt perturbate. Potrivit geneticienilor din programul de cercetare Genomului Uman, aceste procese duc la o reducere a proliferării celulelor embrionare ale crestei neurale - o creastă de-a lungul șanțului neuronal, care se închide într-un tub neural în timpul dezvoltării embrionare.
Formarea țesuturilor faciale are loc datorită transformării și diferențierii celulelor din partea superioară (a capului) a crestei neurale, care migrează de-a lungul tubului neural către zona primului și celui de-al doilea arc ramial al embrionului. Iar deficiența acestor celule provoacă deformări craniofaciale. Perioada critică pentru apariția anomaliilor este de la 18 la 28 de zile după fertilizare. La finalizarea migrării celulelor crestei neurale (în a patra săptămână de gestație), se formează aproape toate țesuturile mezenchimale laxe din zona facială, care ulterior (de la 5 la 8 săptămâni) se diferențiază în țesuturi scheletice și conjunctive ale tuturor părților feței, gâtului, laringelui, urechii (inclusiv urechea internă) și viitorilor dinți.
Patogeneza
Patogeneza sindromului Treacher Collins este adesea familială, iar anomalia este moștenită autosomal dominant, deși există cazuri de transmitere autosomal recesivă a defectului (cu mutații în alte gene, în special POLR1C și POLR1D). Cel mai imprevizibil lucru legat de disostoza maxilo-facială este că mutația este moștenită de copii doar în 40-48% din cazuri. Adică, la 52-60% dintre pacienți, cauzele sindromului Treacher Collins nu sunt asociate cu prezența unei anomalii în familie și se crede că patologia apare ca urmare a unor mutații genetice sporadice de novo. Cel mai probabil, noile mutații sunt consecințele efectelor teratogene asupra fătului în timpul sarcinii.
Printre cauzele teratogene ale acestui sindrom, experții menționează doze mari de etanol (alcool etilic), radiațiile, fumul de țigară, citomegavirusul și toxoplasma, precum și erbicidele pe bază de glifosat (Roundal, Glyfor, Tornado etc.). Iar lista factorilor iatrogeni include medicamente pentru acnee și seboree cu acid 13-cis-retinoic (Isotretinoin, Accutane); medicamentul anticonvulsivant Fenitoina (Dilantin, Epanutin); medicamente psihotrope Diazepam, Valium, Relanium, Seduxen.
Simptome Sindromul Treacher Collins
În cea mai mare parte, semnele clinice ale disostozei mandibulofasciale și gradul de expresie a acestora depind de caracteristicile manifestării mutațiilor genetice. Iar primele semne ale acestei anomalii sunt vizibile în majoritatea cazurilor la un copil imediat după naștere: fața cu sindromul Treacher Collins are un aspect caracteristic. Mai mult, anomaliile morfologice sunt de obicei bilaterale și simetrice.
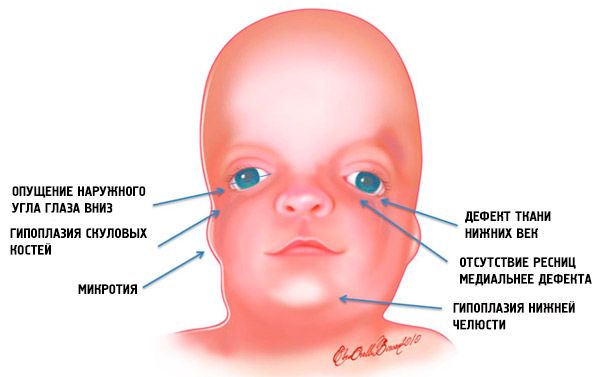
Cele mai evidente simptome ale sindromului Treacher Collins sunt:
- subdezvoltarea (hipoplazia) oaselor faciale ale craniului: zigomatice, procese zigomatice ale osului frontal, plăci pterigoide laterale, sinusuri paranazale, maxilar inferior și proeminențe ale epifizelor osoase (condili);
- subdezvoltarea oaselor maxilarului inferior (micrognație) și un unghi mandibular mai obtuz decât de obicei;
- nasul are dimensiuni normale, dar apare mare din cauza hipoplaziei arcadelor superciliare și a subdezvoltării sau absenței arcadelor zigomatice din regiunea temporală;
- fantele ochilor sunt în jos, adică forma ochilor este anormală, cu colțurile exterioare alungate în jos;
- defecte ale pleoapelor inferioare (colobom) și absența parțială a genelor pe acestea;
- auricule de formă neregulată cu o gamă largă de abateri, inclusiv localizarea lor în colțul maxilarului inferior, absența lobilor, fistule oarbe între tragusul urechii și colțul gurii etc.;
- îngustarea sau închiderea (atezie) canalului auditiv extern și anomalii ale oscioarelor urechii medii;
- absența sau hipoplazia glandelor salivare parotide;
- hipoplazie faringiană (îngustarea faringelui și a căilor respiratorii);
- nefuziunea palatului dur (palatoschizis), precum și absența, scurtarea sau imobilitatea palatului moale.
Astfel de anomalii anatomice au în toate cazurile complicații. Acestea sunt tulburări funcționale de auz sub formă de pierdere a auzului conductiv sau surditate completă; deficiențe de vedere datorate formării incorecte a globilor oculari; defecte ale palatului care cauzează dificultăți la hrănire și înghițire. Există tulburări de ocluzie dentară (malocluzie) asociate cu defecte ale maxilarului, care, la rândul lor, cauzează probleme de mestecare și articulare. Patologiile palatului moale explică vocea nazală.
Complicații și consecințe
Consecințele anomaliilor maxilo-faciale în sindromul Treacher Collins sunt că la naștere abilitățile intelectuale ale copilului sunt normale, dar din cauza defectelor de auz și a altor tulburări, se observă retard mintal secundar.
În plus, copiii cu astfel de defecte își simt acut inferioritatea și suferă, ceea ce le afectează negativ sistemul nervos și psihicul.
Diagnostice Sindromul Treacher Collins
Diagnosticul postnatal al sindromului Treacher Collins se bazează în esență pe semnele clinice. Disostoza craniofacială este ușor de identificat atunci când sindromul este pe deplin expresiv, dar când sunt prezente simptome minim exprimate ale patologiei, pot apărea probleme cu stabilirea diagnosticului corect.
În acest caz, trebuie acordată o atenție deosebită evaluării tuturor funcțiilor asociate cu anomaliile, în special a celor care afectează respirația (din cauza riscului de apnee în somn). De asemenea, trebuie evaluate și monitorizate eficacitatea hrănirii și saturația oxigenului din hemoglobină.
Mai târziu, în a 5-a-6-a zi după naștere, va trebui determinată amploarea afectării auzului prin intermediul unor teste audiologice, care ar trebui efectuate în maternitate.
Se prescrie un examen, în timpul căruia se efectuează diagnostice instrumentale prin fluoroscopie a dismorfologiei craniofaciale; pantomografie (radiografie panoramică a structurilor osoase ale craniului facial); tomografie computerizată craniană completă în diverse proiecții; CT sau RMN al creierului pentru a determina starea canalului auditiv intern.
Cel mai timpuriu diagnostic – prenatal – al anomaliilor maxilo-faciale în prezența sindromului Treacher Collins în antecedentele familiale este posibil prin biopsie de vilus corionic la 10-11 săptămâni de sarcină (procedura amenință avortul spontan și infectarea uterului).
De asemenea, se fac analize de sânge de la membrii familiei; la 16-17 săptămâni de sarcină, se analizează lichidul amniotic (amniocenteză transabdominală); la 18-20 săptămâni de sarcină, se efectuează fetoscopia și se recoltează sânge din vasele fetale ale placentei.
Dar cel mai adesea, ecografia este utilizată în diagnosticul prenatal al acestui sindrom la făt (la 20-24 săptămâni de sarcină).
Ce teste sunt necesare?
Diagnostic diferentiat
Aceleași metode sunt utilizate de specialiști atunci când este nevoie de diagnostic diferențial pentru a recunoaște sindromul Treacher Collins ușor și a-l distinge de alte anomalii congenitale ale oaselor craniofaciale, în special: sindroamele Apert, Crouzon, Nager, Peters-Hewels, Hellermann-Steph, precum și microsomia hemifacială (sindromul Goldenhar), hipertelorismul, fuziunea prematură a suturilor craniene (craniosinostoză) sau fuziunea afectată a oaselor faciale (craniosinostoză).
Cine să contactați?
Tratament Sindromul Treacher Collins
Ca în toate cazurile de defecte congenitale determinate genetic, tratamentul formelor severe de sindrom Treacher Collins este exclusiv paliativ, deoarece pur și simplu nu există metode terapeutice pentru astfel de patologii. Spectrul și gradul deformărilor în acest sindrom sunt extinse și, prin urmare, natura și intensitatea intervenției medicale prezintă, de asemenea, numeroase opțiuni.
Aparatele auditive sunt folosite pentru corectarea și îmbunătățirea auzului, iar ședințele de logopedie sunt folosite pentru îmbunătățirea vorbirii.
Intervențiile chirurgicale sunt necesare la o vârstă fragedă în cazurile severe de îngustare a căilor respiratorii (se efectuează o traheostomie) și a laringelui (se efectuează o gastrostomie pentru hrănire). De asemenea, poate fi necesară corectarea chirurgicală a palatului.
Intervențiile chirurgicale de alungire a mandibulei se efectuează la vârsta de 2-3 ani sau mai târziu. Reconstrucția țesuturilor moi include corectarea colobomului pleoapei inferioare și chirurgia plastică auriculară.
Profilaxie
Prognoză
Care este prognosticul acestei patologii? Depinde de gradul de deformare și de intensitatea simptomelor. Sindromul Treacher Collins este un diagnostic pe tot parcursul vieții.
 [ 25 ]
[ 25 ]