
Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Lissencefalia creierului
Expert medical al articolului

Ultima examinare: 12.07.2025
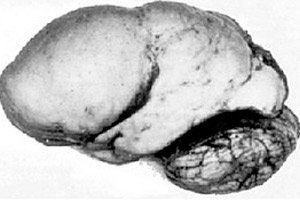
Printre patologiile cerebrale organice, se remarcă o astfel de anomalie congenitală a dezvoltării creierului precum lisencefalia, a cărei esență constă în suprafața aproape netedă a cortexului emisferelor sale - cu un număr insuficient de circumvoluții și șanțuri. [ 1 ]
În absența completă a circumvoluțiilor, se definește agiria, iar prezența mai multor circumvoluții plate și late se numește pahigirie. Aceste defecte, ca și alte deformări de reducere a creierului, au codul Q04.3 în ICD-10.
Epidemiologie
Conform statisticilor privind bolile rare, există 1-1,2 cazuri de lisencefalie la 100 de mii de nou-născuți. [ 2 ], [ 3 ]
Conform unor date, până la 25-30% din cazurile de lisencefalie clasică sunt observate la copiii cu sindrom Miller-Dieker; mutații punctuale și deleții ale genelor LIS1 și DCX sunt detectate la aproape 85% dintre pacienți. [ 4 ]
Studiile genetice efectuate pe 17 gene asociate cu lisencefalia au arătat că mutația sau deleția LIS1 este reprezentată de 40% dintre pacienți, iar 23% sunt asociate cu mutația DCX, urmate de TUBA1A (5%) și DYNC1H1 (3%).[ 5 ]
Cauze lizencefalie
Toate cauzele cunoscute ale formării cortexului cerebral (cortex cerebri) aproape sau complet fără circumvoluții și șanțuri, care măresc „zona de lucru” a creierului uman și asigură „productivitatea” sistemului nervos central, sunt asociate cu tulburări ale dezvoltării sale perinatale. Adică, lisencefalia se dezvoltă la făt. [ 6 ]
Eșecul de a forma straturi ale cortexului cerebral al fătului în lisencefalie este rezultatul migrării anormale a neuronilor care îl formează sau al încetării premature a acestui proces.
Acest proces, esențial pentru histogeneza cerebrocorticală, are loc în mai multe etape, de la săptămâna a 7-a până la a 18-a de sarcină. Și, având în vedere sensibilitatea sa crescută la mutațiile genetice, precum și la diverse influențe fizice, chimice și biologice negative, orice abatere de la normă poate duce la localizarea incorectă a neuronilor, cu posibila formare a unui strat îngroșat de substanță cenușie a cortexului, fără o structură caracteristică. [ 7 ]
În unele cazuri, lisencefalia la copii este asociată cu sindroamele Miller-Dieker, Walker-Warburg sau Norman-Roberts.
Citește și – Defecte de dezvoltare ale creierului
Factori de risc
Pe lângă mutațiile unor gene, factorii de risc pentru nașterea unui copil cu un astfel de defect sever includ privarea de oxigen (hipoxia) a fătului; alimentarea insuficientă cu sânge a creierului (hipoperfuzie); accidentul cerebrovascular acut sub formă de accident vascular cerebral perinatal; patologiile placentare; infecțiile virale ale femeii însărcinate (inclusiv TORCH); [ 8 ] probleme cu metabolismul general și funcția tiroidiană; fumatul, alcoolul, substanțele psihotrope și narcotice; utilizarea unui număr de medicamente; niveluri crescute de radiații. [ 9 ]
Patogeneza
Nu toate cazurile de lisencefalie au o patogeneză cauzată de anomalii cromozomiale și mutații genetice. Însă sunt cunoscute unele gene care codifică proteine ce joacă un rol important în mișcarea corectă a neuroblastelor și neuronilor de-a lungul celulelor gliale radiale - pentru a forma cortexul cerebral. Iar mutațiile acestor gene duc la această patologie. [ 10 ]
În special, acestea sunt mutații sporadice (fără ereditate) ale genei LIS1 de pe cromozomul 17, care reglează proteina motorie citoplasmatică a microtubulilor dineină, precum și gena DCX de pe cromozomul X, care codifică proteina dublecortină (lissencefalină-X). [ 11 ]. În primul caz, specialiștii definesc lissencefalia clasică (tipul I), în al doilea – legată de X. [ 12 ]
Când gena FLN1, care codifică fosfoproteina filamină 1, este deletă, procesul de migrare neuronală direcționată s-ar putea să nu înceapă deloc, ceea ce duce la absența completă a convoluțiilor (agiriei). [ 13 ]
Au fost identificate mutații în gena CDK5, care codifică o enzimă kinază – un catalizator pentru metabolismul intracelular, care reglează ciclul celular în neuronii SNC și asigură migrarea lor normală în timpul formării prenatale a structurilor cerebrale.
Modificările anormale ale genei RELN de pe cromozomul 7 care cauzează defecte girale corticale în sindromul Norman-Roberts duc la o deficiență a glicoproteinei extracelulare reelină, care este necesară pentru reglarea migrației și poziționării celulelor stem neuronale în timpul dezvoltării cortexului cerebral.[ 14 ], [ 15 ], [ 16 ]
Gena ARX codifică proteina homeobox non-aristalens, un factor de transcripție care joacă un rol important în prozencefal și în alte țesuturi.[ 17 ] Copiii cu mutația ARX prezintă și alte simptome, cum ar fi lipsa unor părți ale creierului (agenezie a corpului calos), anomalii ale organelor genitale și epilepsie severă.[ 18 ],[ 19 ]
Mai multe gene au fost asociate cu lisencefalia. Printre acestea se numără VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A și CDK5.[ 20 ]
Citomegalovirusul (CMV) a fost asociat cu dezvoltarea lisencefaliei din cauza scăderii aportului de sânge la creierul fetal. Severitatea infecției cu CMV depinde de vârsta gestațională. Infecția precoce este mai probabil să provoace lisencefalie deoarece migrarea neuronală are loc la începutul sarcinii.[ 21 ]
În plus, mecanismul de apariție al acestei anomalii include încetarea incompletă sau ulterioară a migrării neuronilor din zona generativă periventriculară către cortexul cerebral. Și în astfel de cazuri, se dezvoltă fie lisencefalie incompletă, fie pahigirie, în care se formează mai multe șanțuri și circumvoluții largi (dar majoritatea lipsesc).
Simptome lizencefalie
Primele semne ale acestei patologii (în absența sindroamelor menționate anterior) pot să nu apară imediat după naștere, ci după o lună și jumătate până la două luni. Și cel mai adesea, se observă următoarele simptome clinice ale lisencefaliei:
- hipotonie musculară, adesea asociată cu paralizie spastică;
- convulsii și crize tonico-clonice generalizate (sub formă de opistotonus);
- retard mintal sever și retard de creștere;
- afectarea funcțiilor neurologice și motorii.
Problemele de înghițire cauzează dificultăți în hrănirea bebelușului. [ 22 ]
Un grad ridicat de afectare neuromotorie se manifestă adesea ca tetraplegie - paralizie a tuturor membrelor. Este posibilă deformarea mâinilor, degetelor de la mâini sau de la picioare.
În sindromul Norman-Roberts cu lisencefalie de tip I, se observă anomalii craniofaciale: microcefalie severă, frunte joasă și punte nazală lată și proeminentă, ochi depărtați (hiperterlorism), subdezvoltare a maxilarelor (micrognație). [ 23 ]
Sindromul Miller-Dieker poate fi caracterizat și printr-o dimensiune anormal de mică a capului, cu o frunte lată și înaltă și un nas scurt, depresiuni în tâmple (depresiuni bitemporale) și urechi deformate și implantate jos.
Sindromul de lisencefalie severă este caracterizat prin microcefalie, o scădere a dimensiunii globilor oculari (microftalmie), combinată cu displazie retiniană, hidrocefalie obstructivă și corp calos absent sau hipoplazic.
Complicații și consecințe
Printre complicațiile acestei anomalii, specialiștii numesc o încălcare a funcției de înghițire (disfagie) și reflux gastroesofagian; epilepsie refractară (necontrolată); infecții frecvente ale tractului respirator superior; pneumonie (inclusiv aspirație cronică).
Sugarii cu lisencefalie pot avea probleme cardiace organice congenitale sub forma unui defect septal atrial sau a unui defect cardiac complex cu cianoză (tetralogia Fallot). [ 24 ]
Consecințele retardului de creștere postnatală duc, în majoritatea cazurilor, la deces în decurs de 24 de luni de la naștere.
Diagnostice lizencefalie
Diagnosticul începe cu un examen fizic al copilului, un studiu al istoricului medical al părinților și istoricul sarcinii și al nașterii.
În timpul gestației, pot fi necesare testarea ADN-ului fetal fără celule, amniocenteza sau prelevarea de biopsie de vilus corionic. [ 25 ] Pentru mai multe informații, consultați – Diagnosticul prenatal al bolilor congenitale
Diagnosticarea instrumentală este utilizată pentru vizualizarea structurilor cerebrale și evaluarea funcțiilor acestora:
- tomografie computerizată a creierului;
- imagistica prin rezonanță magnetică (IRM) a creierului;
- electroencefalogramă (EEG). [ 26 ]
În timpul sarcinii, lisencefalia la ecografia fătului după 20-21 de săptămâni poate fi suspectată în absența șanțurilor parieto-occipitale și calcarine și a unei anomalii a fisurii silviene a creierului.
Diagnostic diferentiat
Se efectuează diagnosticul diferențial cu alte sindroame ale defectelor cerebrale congenitale.
Există peste 20 de tipuri de lisencefalie, majoritatea încadrându-se în 2 categorii principale: lisencefalie clasică (tipul 1) și lisencefalie cobblestone (tipul 2). Fiecare categorie are manifestări clinice similare, dar mutații genetice diferite.[ 27 ]
Examinarea creierului în lisencefalia de tip I arată un cortex cerebral cu patru straturi în loc de șase observate la pacienții normali, în timp ce în lisencefalia de tip 2 cortexul cerebral este dezorganizat și apare nodular sau neuniform din cauza deplasării complete a cortexului cerebral de către grupuri de neuroni corticali separați de țesut gliomezenchimal. Pacienții au prezentat, de asemenea, anomalii musculare și oculare.
- Lisencefalie clasică (tipul 1):
- LIS1: Lisencefalie izolată și sindrom Miller-Dieker (lisencefalie asociată cu dismorfism facial). [ 28 ]
- LISX1: Mutație a genei DCX. Comparativ cu lissencefalia cauzată de mutațiile LIS1, DCX prezintă un cortex cu șase straturi în loc de patru.
- Lisencefalie izolată fără alte defecte genetice cunoscute
- Lisencefalie cu aspect de cobblestone (tipul 2):
- Sindromul Walker-Warburg
- Sindromul Fukuyama
- Boli ale mușchilor, ochilor și creierului
- Alte tipuri nu pot fi plasate într-una dintre cele două grupe de mai sus:
- LIS2: Sindromul Norman-Roberts, similar cu lissencefalia de tip I sau sindromul Miller-Dieker, dar fără deleția cromozomului 17.
- LIS3
- LISX2
Microlisencefalie: Aceasta este o combinație între absența plierii corticale normale și un cap anormal de mic. Copiii cu lisencefalie regulată au capete de dimensiuni normale la naștere. Copiii cu un cap mic la naștere sunt de obicei diagnosticați cu microlisencefalie.
De asemenea, este important să se facă distincția între lisencefalie și polimicrogirie, care sunt defecte diferite de dezvoltare a creierului.
Cine să contactați?
Tratament lizencefalie
Lisencefalia este un defect organic incurabil, așadar este posibil doar un tratament suportiv și simptomatic. [ 29 ]
În primul rând, este vorba despre utilizarea anticonvulsivantelor și a medicamentelor antiepileptice, precum și despre instalarea unui tub de gastrostomie în stomac (dacă copilul nu este capabil să înghită singur). Masajul este util.
În cazurile de hidrocefalie severă, se elimină lichidul cefalorahidian.
Profilaxie
Experții recomandă viitorilor părinți să solicite consiliere genetică și ca femeile însărcinate să se înregistreze la medicii obstetricieni și ginecologi în timp util și să se supună tuturor examinărilor programate.
Prognoză
Pentru copiii cu lisencefalie, prognosticul depinde de gradul acesteia, dar cel mai adesea dezvoltarea mentală a copilului nu depășește nivelul de patru până la cinci luni. Și toți copiii cu acest diagnostic suferă de tulburări psihomotorii severe și epilepsie dificil de tratat. [ 30 ]
Conform NINDS (Institutul Național American pentru Tulburări Neurologice și Accident Vascular Cerebral), speranța maximă de viață pentru persoanele cu lisencefalie este de aproximativ 10 ani.