
Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Encefalomiopatia Leah necrotizantă subacută
Expert medical al articolului

Ultima examinare: 04.07.2025
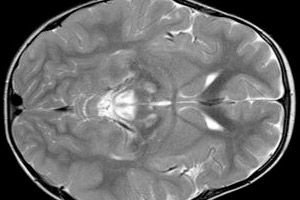
Boala a fost menționată pentru prima dată în 1951. Până în prezent, au fost descrise peste 120 de cazuri. Boala Leigh (OMIM 256000) este o boală genetic eterogenă care poate fi moștenită fie nuclear (autosomal recesiv sau legată de cromozomul X), fie mitocondrial (mai puțin frecventă).
 [
[ Cauze a sindromului Leah
Boala are la bază un deficit de enzime care asigură producția de energie în principal din cauza unei perturbări a metabolismului acidului piruvic și a unui defect în transportul electronilor în lanțul respirator. Se dezvoltă un deficit al complexului piruvat dehidrogenazei (subunitatea α-E1), al piruvat carboxilazei, al complexului 1 (NAD-coenzima Q-reductază) și al complexului 4 (citocrom oxidază) din lanțul respirator.
S-a stabilit că defectele piruvat carboxilazei, complexului 1 (NAD-coenzima Q-reductază) și complexului 4 (citocrom oxidază) din lanțul respirator sunt moștenite autosomal recesiv, defectele complexului piruvat dehidrogenazei (subunitatea a-E1) sunt moștenite recesiv legat de cromozom X. În cazul mutațiilor punctuale ale ADNmt, care afectează a 6-a subunitate a ATP-azei, moștenirea mitocondrială este tipică. Cel mai adesea, apare o mutație miscens, asociată cu înlocuirea timinei cu guanină sau citozină la poziția 8993 a ADNmt. Mai puțin frecventă este o mutație la poziția 9176 a ADNmt. Datorită faptului că mutația T8993G este principalul defect în sindromul NARP, au fost descrise familii cu aceste două boli. La copii, a fost descrisă și o mutație în ADNmt la poziția 8344, care apare în sindromul MERRF.
Se presupune că, în cazul acumulării de ADNmt mutant în majoritatea mitocondriilor, se dezvoltă o evoluție severă a sindromului Leigh. În geneza mitocondrială a acestei afecțiuni, ADNmt mutant se găsește în 90% din totalul mitocondriilor. Patogeneza este asociată cu o încălcare a formării energiei în celule și cu dezvoltarea acidozei lactice.
Simptome a sindromului Leah
Primele semne ale bolii debutează la o vârstă fragedă (1-3 ani). Cu toate acestea, există cazuri cunoscute de manifestare a bolii la 2 săptămâni și la 6-7 ani. La început, se dezvoltă tulburări nespecifice: dezvoltare psihomotorie întârziată, scăderea poftei de mâncare, episoade de vărsături, deficit de greutate corporală. Ulterior, simptomele neurologice se intensifică: hipotonie musculară sau distonie cu trecere la hipertonie, atacuri de mioclonie sau convulsii tonico-clonice, tremor al extremităților, coreoatetoză, tulburări de coordonare, scăderea reflexelor tendinoase, letargie, somnolență. Neurodegenerarea cerebrală este progresivă. Simptomele insuficienței piramidale și extrapiramidale se intensifică, actul deglutiției este afectat. Se observă adesea modificări ale organului vederii precum ptoză, oftalmoplegie, atrofie a nervilor optici, mai rar degenerare pigmentară a retinei. Uneori se dezvoltă cardiomiopatie hipertrofică, apar episoade de tahipnee.
Rareori, boala evoluează ca o encefalopatie acută. Mai tipică este o evoluție cronică sau subacută, care duce la un rezultat fatal la câțiva ani de la debutul bolii. Cu o evoluție rapidă (câteva săptămâni), moartea survine ca urmare a paraliziei centrului respirator.
Diagnostice a sindromului Leah
Un test biochimic de sânge relevă acidoza lactică datorată acumulării de acizi lactici și piruvici în sânge și lichidul cefalorahidian, precum și o creștere a conținutului de alanină în sânge. Nivelul corpilor cetonici poate fi, de asemenea, crescut. În urină se detectează o excreție crescută de acizi organici: lactic, fumaric etc. Nivelul de carnitină din sânge și țesuturi scade adesea.
Rezultatele EEG relevă semne focale de activitate epileptică. Datele RMN relevă mărirea ventriculelor cerebrale, leziuni cerebrale bilaterale, calcificarea ganglionilor bazali (nucleul caudat, putamen, substantia nigra, globus pallidus). De asemenea, poate fi detectată atrofia emisferelor cerebrale și a materiei cerebrale.
Examinarea morfologică relevă modificări macroscopice ale materiei cerebrale: focare simetrice de necroză, demielinizare și degenerare spongioasă a creierului, în principal a secțiunilor medii, pontului, ganglionilor bazali, talamusului și nervului optic. Tabloul histologic include degenerarea chistică a țesutului cerebral, glioza astrocitară, moartea neuronală și o creștere a numărului de mitocondrii din celule. În mușchii scheletici se observă acumularea de incluziuni lipidice, o scădere a reacției histochimice la complexele 1 și 4 ale lanțului respirator, acumulare subsarcolemală de mitocondrii, mitocondrii anormale cu dezorganizarea crestelor. Fenomenul de RRF adesea nu este detectat.
Cum să examinăm?
Ce teste sunt necesare?