
Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Sindromul Aper
Expert medical al articolului

Ultima examinare: 04.07.2025

 [
[ Cauze Sindromul Aper
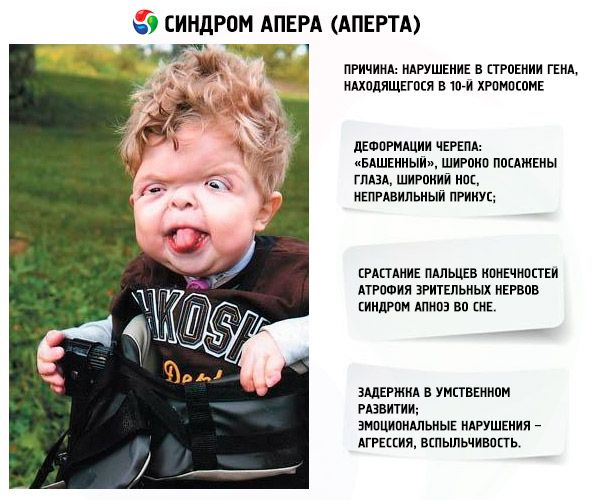
Dezvoltarea sindromului este provocată de o tulburare a structurii unei gene situate pe cromozomul 10. Această genă este responsabilă de procesul de separare a degetelor și, în plus, de închiderea la timp a suturilor craniene.
În plus, bolile infecțioase suferite în timpul sarcinii (cum ar fi rubeola sau tuberculoza, sifilisul sau gripa, precum și meningita) și expunerea mamei la raze X sunt considerate cauze ale patologiei. Cel mai adesea, un astfel de sindrom este detectat la copiii născuți din părinți vârstnici.
Patogeneza
Sindromul Apert este ereditar. Tipul său este autosomal dominant (adică, într-o familie în care unul dintre viitorii părinți este bolnav, probabilitatea de a avea un copil cu această boală este de 50-100%).
O mutație unică a receptorului 2 al factorului de creștere a fibroblastelor (FGFR2) are ca rezultat un număr crescut de celule progenitoare care se dezvoltă de-a lungul căii osteogenice. Acest lucru duce în cele din urmă la creșterea formării matricei osoase subperiostale și la osificarea prematură a suturilor craniene în timpul dezvoltării fetale. Ordinea și rata de topire a suturilor determină gradul de deformare și dizabilitate. Odată ce materialul de sutură s-a vindecat, creșterea altor țesuturi perpendiculare pe această sutură este limitată, iar oasele fuzionate acționează ca o singură schelă osoasă.
Prima dovadă genetică că sindactilia în sindromul Apert se datorează unui defect al receptorului factorului de creștere al keratinocitelor (KGFR) a fost observarea unei corelații între expresia KGFR în fibroblaste și severitatea sindactiliei.
Ambliopia și strabismul sunt mai frecvente la pacienții cu mutația FGFR2 Ser252Trp, iar atrofia discului optic este mai frecventă la pacienții cu mutația FGFR2 Pro253Arg. Pacienții cu mutații FGR2 Ser252Trp au o prevalență semnificativ mai mare a deficienței de vedere comparativ cu pacienții cu mutația FGFR2 Pro253Arg.
Simptome Sindromul Aper
Unele manifestări ale bolii sunt clar vizibile deja la nașterea bebelușului, deoarece se dezvoltă în uterul mamei. Printre principalele simptome ale sindromului:
- Craniul este deformat - se întinde în înălțime, dobândind un aspect de „turn”; în plus, ochii sunt amplasați larg și ușor bombați (deoarece dimensiunea orbitelor scade); nasul se lărgește și se formează o mușcătură incorectă (dinții superiori ies excesiv în afară);
- Degetele membrelor sunt complet fuzionate (în principal degetul inelar, precum și degetul mijlociu și degetul arătător) și arată ca o membrană a pielii sau o fuziune completă a oaselor; în plus, pot crește degete suplimentare;
- Retardare mintală (nu apare la toată lumea);
- Nervii optici se atrofiază, ca urmare a scăderii acuității vizuale (în unele cazuri, se dezvoltă pierderea completă a vederii);
- Creșterea presiunii intracraniene, care apare ca urmare a închiderii premature a suturilor craniene, se manifestă sub formă de dureri de cap și vărsături cu greață;
- Deoarece maxilarul superior rămâne subdezvoltat, apar probleme respiratorii;
- Sindromul de apnee în somn este o manifestare frecventă.
- Manifestări emoționale – agresivitate, lipsă de reținere, irascibilitate puternică.
Complicații și consecințe
Majoritatea pacienților prezintă un anumit grad de obstrucție a căilor respiratorii superioare în copilărie. Căile respiratorii superioare sunt slab patentate din cauza dimensiunii reduse a nazofaringelui și a coanei, iar căile respiratorii inferioare pot cauza moarte prematură din cauza anomaliilor cartilajului traheal.
Diagnostice Sindromul Aper
Pentru a stabili un diagnostic, sunt necesari următorii pași:
- Medicul ar trebui să analizeze plângerile pacientului, precum și istoricul medical. Este necesar să se afle dacă au existat cazuri de patologie similară în familie;
- Examen neurologic pentru evaluarea formei craniului și a dezvoltării intelectuale a pacientului (chestionare speciale, precum și interviu);
- Examinarea fundului de ochi pentru a detecta prezența simptomelor de creștere a presiunii intracraniene (umflarea discului optic, precum și estomparea marginilor acestuia);
- Pentru a evalua starea craniului, se efectuează o radiografie;
- Imagistică computerizată și prin rezonanță magnetică a capului pentru a examina structura creierului și a craniului strat cu strat, pentru a determina prezența simptomelor de fuziune prematură a suturilor craniene și, în plus, hidrocefalie (din cauza creșterii presiunii intracraniene, se acumulează exces de lichid cefalorahidian (acesta este lichidul cefalorahidian care promovează procesul metabolic, precum și nutriția creierului));
- Radiografie a picioarelor și mâinilor pentru a determina cauza fuziunii degetelor (acest lucru este important pentru planificarea intervenției chirurgicale ulterioare);
- Se poate prescrie o consultație cu un genetician medical și un neurochirurg.
Teste
Se efectuează analiza genetică a mutațiilor comune care apar în gena de tip FGFR2.
Diagnostic diferentiat
Acest sindrom trebuie diferențiat de alte patologii genetice în care se observă craniosinostoză. Acestea sunt boli precum sindroamele Pfeiffer, Crouzon, Saethre-Chotzen și Carpenter. Metodele de testare genetică moleculară sunt utilizate pentru a exclude aceste anomalii.
Cine să contactați?
Tratament Sindromul Aper
Chirurgia este considerată singurul tratament eficient pentru sindromul Apert – aceasta ajută la corectarea defectelor fizice individuale și corectează, de asemenea, retardul mintal.
În timpul acestei proceduri, sutura coronală este închisă pentru a preveni posibile leziuni cerebrale. Cea mai comună tehnică este distragerea craniofacială, care implică tracțiunea gradată a craniului. Chirurgia ortodontică și/sau ortognatică se efectuează pentru a îndepărta defectele faciale individuale.
În plus, pacienții sunt supuși unei corecții chirurgicale a fuziunii degetelor.
Referințe
Genetica medicala. Ghid național. Editat de Ginter EK, Puzyrev VP GEOTAR-Media. 2022