
Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Idioțenie amaurotică
Expert medical al articolului

Ultima examinare: 04.07.2025

Idiocia amaurotică este o boală progresivă rară. Se caracterizează printr-o scădere treptată a vederii până la orbire completă și degradarea inteligenței până la instalarea idioției. Drept urmare, pacientul dezvoltă un marasm profund cu rezultat fatal. Boala a fost descrisă pentru prima dată de oftalmologul Dr. Tau în urmă cu mai bine de 130 de ani. Tau a observat o transformare specială a fundului de ochi. Peste 500 de cazuri ale acestei boli au fost deja descrise.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
Epidemiologie
Epidemiologia bolii remarcă o natură familială. În familiile în care există pacienți cu idioție amaurotică, căsătoriile consangvine sunt periculoase din același motiv. De asemenea, sunt expuse riscului persoanele de origine canadiano-franceză sau evreiască.
[ 11 ], [ 12 ], [ 13 ], [ 14 ], [ 15 ], [ 16 ], [ 17 ], [ 18 ]
Cauze idioțenie amaurotică
În ciuda numeroaselor date colectate despre boală, oamenii de știință continuă în prezent să caute răspunsuri la multe întrebări despre cauzele, patogeneza și chiar manifestările idioției amaurotice.
Există sugestii conform cărora boala este ereditară. Tipul de moștenire este autosomal recesiv. Cel mai adesea, sunt afectate cerebelul și lobii occipitali ai emisferelor cerebrale, cu consecințe severe și complicații pentru întregul corp: atrofia nervilor optici, fibrele nervoase își pot pierde membranele, iar conexiunile dintre celulele nervoase se pot dezintegra.
Majoritatea experților recunosc că semnele clinice ale bolii pot fi destul de variate și se corelează cu vârsta la care idioția amaurotică a început să se dezvolte la pacient.
În timpul studiului cauzelor bolii, s-a observat un anumit tipar: boala afectează adesea copiii din aceeași familie, motiv pentru care se folosește denumirea de „idioție amaurotică familială”. Conform studiilor, ale căror rezultate au fost publicate la începutul studiului bolii, din 64 de cazuri de idioție amaurotică, 37 au fost găsite în 13 familii (fiecare familie avea 2-5 copii bolnavi). Este demn de remarcat faptul că în astfel de familii, bolnavii aveau frați și surori absolut sănătoși. În zilele noastre, se crede că factorul de moștenire recesivă joacă un rol important în dezvoltarea bolii. Astfel, este posibil să se explice frecvența apariției cazurilor de boală în aceleași familii. Atunci când se analizează factorul ereditar ca o cauză a idiociei amaurotice, nu trebuie să se limiteze la prezența semnelor exprimate clinic în familiile pacienților (atât pe linia ascendentă, cât și pe cea laterală), ci să se țină cont și de cele rudimentare, de exemplu, deviațiile caracteristice în funcționarea aparatului vizual (coroidită familială, distrofie retiniană pigmentară etc.).
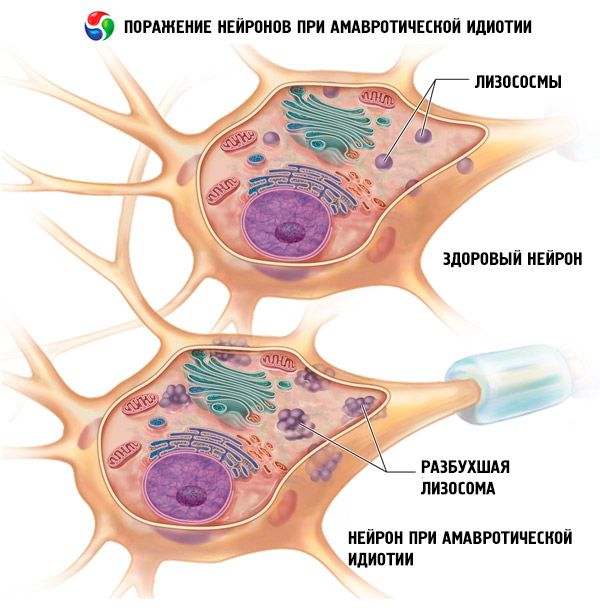
Simptome idioțenie amaurotică
Etape
Forma infantilă se dezvoltă între 4 și 6 luni. Această formă de idiopatie amaurotică este caracterizată printr-o natură familială. Vederea scade rapid: bebelușul nu își poate fixa privirea, nu observă obiectele. Pe fundul de ochi apare așa-numitul „sâmbure de cireșă” - o pată roșiatică în regiunea maculară, care este înconjurată de o margine gri-albă. Apoi, nervii optici se atrofiază, iar copilul își pierde complet capacitatea de a vedea. Orientarea, reflexele de protecție, precum și capacitatea de mișcare se pierd treptat. Pacienții reacționează puternic la stimulii sonori - tresar la un sunet care este liniștit pentru o persoană sănătoasă, se pot observa convulsii din cauza creșterii tonusului muscular. În stadiul final al bolii, se dezvoltă atrofie generală, epuizare a corpului și creșterea tonusului tuturor mușchilor extensori. Prognosticul bolii este, de asemenea, dezamăgitor: pacientul decedează la un an și jumătate până la doi ani de la debutul bolii.
Forma copilăriei târzii începe la 3-4 ani. Boala progresivă alternează cu etape de remisie. Pierderea treptată a inteligenței este însoțită de convulsii, tulburări de coordonare și tulburări extrapiramidale. Această formă este caracterizată și prin atrofia nervului optic. Decesul survine la 6-8 ani de la debutul idiociei amaurotice.
Forma juvenilă începe să se manifeste la 6-10 ani. Idioția amaurotică Spielmeyer progresează mai rar. Modificările fundului de ochi coincid cu manifestările distrofiei retiniene pigmentare. Vederea pacientului scade treptat, la fel și inteligența. Funcțiile motorii afectate se pot manifesta în moduri diferite și inconstant: apar paralizii nu foarte pronunțate ale brațelor și picioarelor, tulburări extrapiramidale și bulbare. Boala duce la deces la 10-25 de ani de la apariția primelor semne.
Forma tardivă apare foarte rar și se dezvoltă extrem de lent. Starea mentală a pacientului se modifică (asemănător unui sindrom mental organic), se observă atrofie a nervilor optici și distrofie pigmentară a retinei. Stadiul final este caracterizat prin paralizie și sindrom epileptiform. Pacientul decedează la 10-15 ani de la debutul bolii.
[ 38 ], [ 39 ], [ 40 ], [ 41 ], [ 42 ], [ 43 ], [ 44 ], [ 45 ]
Formulare
Există patru tipuri de idioție amaurotică:
- Tay-Sachs (afectând la o vârstă fragedă);
- Jansky-Bilynovsky (aparând la copii la o vârstă mai înaintată);
- Sindromul Spielmeyer-Vogt (care apare la adolescenți);
- Kufsa (formă târzie).
Unii oameni de știință disting separat și tipul congenital Norman-Wood.
Fiecare tip de boală are propriul set de manifestări clinice, dar toate sunt unite de cauze comune, tablou clinic, bază anatomică și patogeneză.
[ 46 ], [ 47 ], [ 48 ], [ 49 ], [ 50 ], [ 51 ], [ 52 ], [ 53 ], [ 54 ]
Diagnostice idioțenie amaurotică
Idiocia amaurotică este cauzată de o tulburare a metabolismului lipidic, în urma căreia un produs intermediar al metabolismului lipidic, sfingomielina, se depune în diverse celule ale organismului. Localizarea și compoziția depozitelor determină dezvoltarea unui tablou clinic specific al bolii.
[ 55 ], [ 56 ], [ 57 ], [ 58 ], [ 59 ], [ 60 ], [ 61 ], [ 62 ], [ 63 ]
Cum să examinăm?
Diagnostic diferentiat
Diagnosticul diferențial al idiociei amaurotice se bazează pe un tablou clinic specific și patologiile caracteristice ale fundului de ochi.
Forma incipientă are simptome similare cu boala Landing, un tip de mucopolizaharidoză. Boala Landing se dezvoltă din primele luni după naștere și duce la deces după 2-3 ani. Un „sâmbure de cireșă” pe fundul de ochi apare în 1/5 din cazuri, modificările degenerative ale retinei și percepția distorsionată a sunetelor (hipercuție) nu sunt practic caracteristice acesteia, dar se observă simultan mărirea splinei și a ficatului, tulburări mintale și tulburări de mișcare.
Forma juvenilă se suprapune uneori cu manifestările sindromului Lawrence-Moon-Biedl. Pentru a diferenția aceste boli, este necesar să se acorde atenție și celorlalte manifestări ale acestora. Sindromul Lawrence-Moon-Biedl se caracterizează prin creștere rapidă în greutate, deformarea membrelor caracterizată prin prezența unor degete suplimentare de la mâini sau de la picioare, tulburări vegetative-trofice sesizabile și absența tulburărilor funcției motorii.
Varietatea simptomelor idioției amaurotice tardive complică diagnosticul pe parcursul vieții. Manifestările sale sunt similare cu ataxia Friedreich, scleroza multiplă, boala Alzheimer, boala Pick, paralizia progresivă și chiar schizofrenia.
Unii autori insistă că diagnosticul acestei boli, mai ales când manifestările clinice sunt neclare, poate fi stabilit în mod fiabil doar după deces, pe baza analizei anomaliilor histologice ale sistemului nervos.
Cine să contactați?
Tratament idioțenie amaurotică
Nu există un tratament rațional și eficient. În zilele noastre, terapia pentru idioția amaurotică vizează exclusiv ameliorarea simptomelor. Se utilizează sedative, nootropice, anticonvulsivante și tonice generale.
Pentru a activa circulația sângelui și procesele metabolice din creier, sunt prescrise glicină, elkar, cerebrolizină, acid glutamic și pantogam.
Pentru ameliorarea sindromului convulsiv, se prescriu difenină sau carmazepină.
Un rezultat pozitiv poate fi obținut prin utilizarea extractelor de țesuturi, a transfuziei de sânge sau a plasmei.
Profilaxie
Lipsa unei terapii eficiente pentru idiocia amaurotică ne obligă să acordăm o atenție sporită prevenției. Există deja metode care ne permit să identificăm purtătorii heterozigoți ai genei patologice și metode de diagnosticare a idiociei amaurotice în timpul sarcinii. Diagnosticul prenatal al bolii constă în analiza activității hexosaminidazei A în lichidul amniotic. Dacă se detectează o activitate enzimatică redusă, se recomandă întreruperea sarcinii. Părinții unui copil bolnav sunt sfătuiți să înceteze să mai aibă copii.